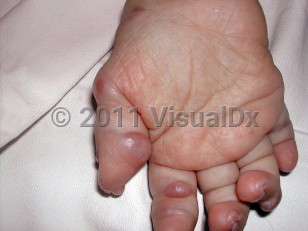
Hyaline fibromatosis syndrome (HFS) is a family of rare genetic syndromes characterized by dysfunctional deposition of hyaline extracellular matrix materials in various tissues, resulting in papulonodular cutaneous lesions, gingival hypertrophy, joint contractures, and osteolytic lesions in infancy or early childhood. In very severe cases, there may be hyaline deposition in a variety of organ systems as well, such as the gastrointestinal tract.
HFS is extremely rare, with fewer than 100 cases reported worldwide, and affects both females and males with equal frequencies. Most of the reported cases have been in people with Turkish, Indian, and Moroccan ethnicity, but the true distribution is unknown.
HFS is the unifying designation that includes juvenile hyaline fibromatosis (JHF) and infantile systemic hyalinosis (ISH). Both disorders are caused by distinct deleterious mutations in the gene encoding for capillary morphogenesis protein 2 (CMG2; also known as anthrax toxin receptor 2 or ANTXR2) and are inherited in an autosomal recessive manner. CMG2 is a transmembrane protein that interacts with proteins in the extracellular matrix, such as collagen VI. It is hypothesized that CMG2 mediates lysosomal transport of collagen VI for degradation, and so mutations in CMG2 lead to the aberrant deposition and accumulation of collagen VI-containing extracellular hyaline material in various tissues.
The clinical presentation of ISH is similar to JHF but is more severe and occurs earlier, in infancy rather than childhood. It has been hypothesized that ISH mutations in CMG2 tend to cause protein truncations whereas JHF mutations tend to be missense mutations that might only mildly impact the function of the resulting protein.
The characteristic cutaneous lesions of HFS are asymptomatic and slow growing, commonly presenting as pink papules around the nose or nodules on the head, back, and knees. Gingival hypertrophy may be severe enough to interfere with feeding, leading to malnutrition and recurrent infections in infancy. The most debilitating disease features are the progressively painful joint contractures that limit mobility, often of the knees, elbows, fingers, and hip joints. Nodules can also form in internal organs and cause end-organ dysfunction. When this occurs in the intestines, affected individuals can present with severe diarrhea and a protein-losing enteropathy with associated failure to thrive.
HFS is frequently progressive and disabling, causing most adolescents and adults to become bedridden or wheelchair dependent. While individuals with JHF and mild disease manifestation can live into adulthood, those with severe disease often do not survive beyond childhood. The most severe cases of ISH can cause early life-threatening complications such as intractable diarrhea, recurrent infections, and multiorgan system failure and can result in death in early infancy.
A grading system for HFS has been proposed that classifies HFS into grade 1 (mild; limited to skin and gingival involvement), grade 2 (moderate; grade 1 + joint involvement), grade 3 (severe; grade 2 + signs of internal organ involvement), and grade 4 (lethal; grade 3 + signs of clinical decompensation).
Hyaline fibromatosis syndrome in Adult
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
M72.8 – Other fibroblastic disorders
SNOMEDCT:
238861002 – Juvenile hyaline fibromatosis
M72.8 – Other fibroblastic disorders
SNOMEDCT:
238861002 – Juvenile hyaline fibromatosis
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:03/17/2020
Last Updated:01/17/2022
Last Updated:01/17/2022