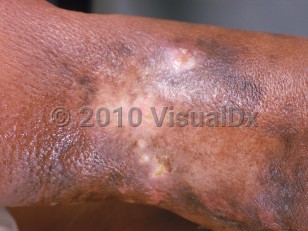
Lymphangiosarcoma (LAS) is a rare, aggressive cutaneous angiosarcoma occurring in association with chronic lymphedema. While it may be associated with various underlying causes of lymphedema (eg, congenital, filarial, or posttraumatic), 90% of cases are seen following mastectomy with lymph node dissection (Stewart-Treves syndrome). Isolated cases of LAS not related to mastectomy yet associated with chronic lymphedema are also reported. Additionally, LAS may also follow local radiation therapy for breast carcinoma.
The term "lymphangiosarcoma" is controversial. While immunohistochemical stains for lymphatic-based antibodies are positive in the majority of cases of Stewart-Treves syndrome and in angiosarcomas induced by radiotherapy, electron microscopy reveals the cells to be surrounded by structures more consistent with a vascular origin (complete basal lamina, intercellular junctions, etc). Use of the term may help distinguish LAS from the more common form of cutaneous angiosarcoma (cAS) that affects the head and neck region of elderly patients. While the exact etiology of LAS is unknown, it is thought that postoperative hematoma formation, chronic infections, tissue fibrosis, and even local radiation may all create a regional immunodeficient state contributing to the development of LAS.
Acute (less than 18 months) and chronic (more than 18 months) lymphedema in the upper extremities occurs in up to 35% of patients following radical mastectomy with axillary lymph node dissection or radiotherapy. LAS has been reported to occur in less than 1% of women at 5 and 15 years postradical mastectomy with lymphedema. Advances in surgical management of breast carcinoma, including partial mastectomies, sentinel lymph node biopsies, and adjuvant radiation therapy, have significantly decreased the incidence of associated postsurgical lymphedema and cases of LAS.
LAS metastases most frequently go to the thoracic cavity and lungs but may also involve other internal organs and bone. Prognosis for LAS is extremely poor, as it is associated with rapid progression and metastases. Therapeutic options include chemotherapy and/or radiation therapy and limb amputation; however, none is considered effective. Five-year survival rates have been reported to be less than 10%.
Pediatric patient considerations: LAS can rarely occur with congenital lymphedema (ie, Milroy disease).
Lymphangiosarcoma
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
C49.10 – Malignant neoplasm of connective and soft tissue of unspecified upper limb, including shoulder
SNOMEDCT:
403986008 – Lymphangiosarcoma
C49.10 – Malignant neoplasm of connective and soft tissue of unspecified upper limb, including shoulder
SNOMEDCT:
403986008 – Lymphangiosarcoma
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:11/01/2021
Last Updated:11/01/2021
Last Updated:11/01/2021