Kindler syndrome (KS) is a rare type of inherited epidermolysis bullosa (EB), which is a group of mechanobullous disorders characterized by defects in the basement membrane zone (BMZ). Instability of the BMZ manifests clinically as skin fragility and bulla formation at sites of trauma.
KS is inherited in an autosomal recessive pattern that results from a loss-of-function in the FERMT1 gene (formerly KIND1), which encodes a protein known as fermitin family homolog 1 (FFH1), a focal adhesion protein that links the actin cytoskeleton with the underlying extra cellular matrix. Weakness at this point causes splits at various levels, most often above the basal keratinocytes.
Like other forms of EB, blisters in KS are typically noted soon after birth or in early infancy; there is a predilection for acral sites. Disease severity may range from mild, with minimal skin disease, to severe with involvement of mucosal sites (such as conjunctiva, vagina, urethra, anus, and mouth) and gastrointestinal tract (causing esophageal strictures and colitis). Patients with mild disease may not be diagnosed until late in adulthood.
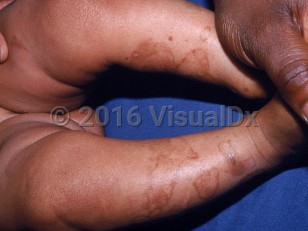
Features that distinguish KS from other types of EB are photosensitivity, progressive poikiloderma, and marked skin atrophy, which begin early in the disease course. While blistering and photosensitivity tend to diminish in severity with age, poikiloderma is permanent. Scarring is usually minimal, but repeated trauma can result in development of scar tissue.
Other clinical findings of KS include hyperkeratosis of the palms and soles, joint laxity, severe periodontal disease, ectropion, webbing of the fingers and toes, nail dystrophy, esophageal and urethral stenosis, and phimosis, although these findings are typically sequelae seen later in childhood or adulthood.
With the increased photosensitivity, there is also an increased risk of nonmelanoma skin cancer in KS, with squamous cell carcinomas (SCC) reported in acral skin and the mouth.
Related topics: EB simplex, generalized severe EB simplex, junctional EB, dystrophic EB, EB acquisita
Kindler syndrome in Infant/Neonate
Alerts and Notices
Important News & Links
Synopsis

Codes
ICD10CM:
Q81.8 – Other epidermolysis bullosa
SNOMEDCT:
238836000 – Kindler syndrome
Q81.8 – Other epidermolysis bullosa
SNOMEDCT:
238836000 – Kindler syndrome
Look For
Subscription Required
Diagnostic Pearls
Subscription Required
Differential Diagnosis & Pitfalls

To perform a comparison, select diagnoses from the classic differential
Subscription Required
Best Tests
Subscription Required
Management Pearls
Subscription Required
Therapy
Subscription Required
References
Subscription Required
Last Reviewed:12/08/2019
Last Updated:01/18/2022
Last Updated:01/18/2022
Kindler syndrome in Infant/Neonate